
Projets émergents
En parallèle des projets développés sur la protéine Tau et la maladie d’Alzheimer, notre équipe intègre d’autres problématiques en lien avec les pathologies neurodégénératives en favorisant le développement de thématiques émergentes et en capitalisant sur nos compétences en neurobiologie fondamentales et cliniques.
Personnels impliqués: Pauline Beauquel, David Blum, Valérie Buée-Scherrer, Baptiste Damary, Claire-Marie Dhaenens, Violette Delforge, Emilie Faivre, Nicolas Geoffre, Vincent Huin, Audrey Penin, Anaïs Poncet, Vasily Smirnov
Développement d’un nouveau modèle sporadique TDP43.
Il n’existe à ce jour aucun traitement curatif pour la Sclérose Latérale Amyotrophique (SLA) et les dégénérescences lobaires fronto-temporales (DLFT), des pathologies neurodégénératives progressives rares ayant des caractéristiques communes mais dont l’étiopathogénie est complexe et pas entièrement comprise. 97% des patients atteints de SLA et 45% des DLFT présentent une accumulation/agrégation cytoplasmique anormale de la protéine TDP-43 (transactive response DNA-binding protein 43) principalement dans les neurones mais également dans les cellules gliales, conduisant aux symptômes moteurs et cognitifs observés dans ces deux pathologies. Néanmoins, la plupart des modèles animaux de pathologies TDP-43 existants, sont basés sur des mutations génétiques et ne reflètent donc pas l’ensemble des cas sporadiques.
Notre projet vise à développer un nouveau modèle sporadique TDP-43 afin de mieux comprendre les mécanismes physiopathologiques impliqués et de tester deux approches thérapeutiques que nous développons dans le centre Lille Neuroscience & Cognition dans la SLA et les DLFT : les biomateriaux plaquettaires et le blocage sélectif des récepteurs adénosinergiques A2A (Collaboration avec Anne-Sophie Rolland de l’équipe TREAT).
Etude des pathologies neurodégénératives liées au gène RFC1.
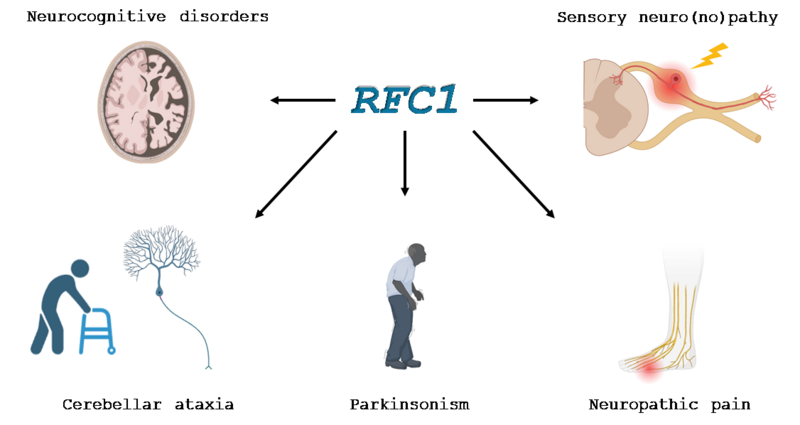
Le CANVAS (Cerebellar ataxia with neuropathy and vestibular areflexia syndrome) est une maladie génétique constituant une cause majeure d'ataxie de début tardif. Cette pathologie est causée par des expansions bialléliques situées dans l’intron 2 du gène RFC1. Ce gène code pour une protéine ubiquitaire, la sous-unité 1 du facteur de réplication C, un complexe protéique impliqué dans la réplication et la réparation de l'ADN des eucaryotes. Il existe une grande hétérogénéité clinique des différentes atteintes dans cette maladie. En effet, cette pathologie se caractérise par une atteinte neurologique complexe et affectant de nombreux éléments du système nerveux avec une neuropathie sensitive presque constante responsable d’une ataxie proprioceptive, une dysautonomie, une atteinte cérébelleuse, une atteinte vestibulaire, une atteinte des motoneurones, des douleurs neuropathiques et une toux chronique. Notre équipe a aussi rapporté un parkinsonisme chez 10 % des patients atteints de CANVAS, une atteinte des motoneurones et de fréquents troubles cognitifs.
Nous nous concentrons sur les aspects cliniques, génétiques et physiopathologiques des pathologies liées à RFC1 avec (i) l’étude des différents endophénotypes de la maladie (Parkinsonisme, atteinte cognitive, douleur…), (ii) l’étude des différentes mutations, et (iii) la compréhension des mécanismes physiopathologiques en abordant l’hypothèse d’une perte de fonction du gène. Pour cela, nous étudions les effets du knock-down du gène RFC1 dans différents modèles cellulaires et animaux avec l’optique de créer de nouveaux modèles de la maladie et d’amorcer le développement de nouvelles stratégies thérapeutiques pour ces maladies liées à RFC1, mais aussi les autres pathologies neurologiques avec des atteintes similaires.
Maladies dégénératives de la rétine.
Nous nous intéressons aux maladies dégénératives de la rétine et en particulier à la découverte de nouveaux gènes impliqués dans les formes rares de dystrophies et de dysfonctions des photorécepteurs cônes ainsi qu’à leur caractérisation phéno- et génotypique. Nous étudions les conséquences cellulaires des variants afin d’identifier des marqueurs pouvant être utiles au diagnostic, au pronostic et au traitement. Nous réalisons également de nombreux tests de validation fonctionnelle des défauts géniques retrouvés dans les dystrophies rétiniennes héréditaires (minigènes, transcriptomique).
Enfin, nous développons des technologies innovantes (long read sequencing) pour étudier des gènes de structure complexe portés par le chromosome X, et pour comprendre notamment l’effet de la méthylation de l’ORF15 de RPGR sur l’expression de la maladie chez les femmes et l’organisation des gènes OPN1LW et OPN1MW codant les opsines des cônes, cause majeure d’anomalies de la vision des couleurs.
Cette thématique met l’accent sur l’impact clinique et la personnalisation des diagnostics avec de nombreuses études de corrélations génotype/phénotype.